These are very prompted by a film. Schizophrenia is an illness that I continue to research in some depth. There is now compelling evidence that schizophrenia is a very complex syndrome that involves numerous neural pathways in the brain, far more than just dopaminergic and serotonergic systems. One of the more popular theories in recent literature is that it represents a hypoglutaminergic deficiency of certain pathways, including thalamic ones.
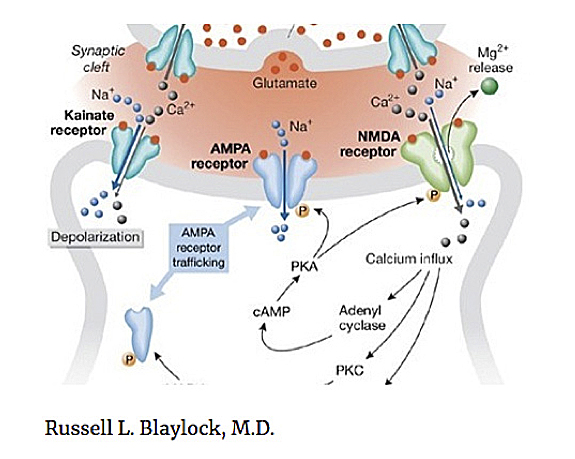
After much review of research and study in this area I have concluded that most such theories are short sighted, and too focused, perhaps looking in the wrong direction. Most are based on clinical responses to certain drugs, particularly antipsychotic drugs affecting the dopaminergic or the glutamate receptor system, mainly the NMDA (M-methyl-D-Aspartame) receptors and its various subunits.
I have written a number of papers on a newer hypothesis of neurodegenerative disease, of which schizophrenia is one. This involves what I have described as immunoexcitotoxicity — that is, an interaction between immune factors and glutamate receptors that leads to degeneration of specific groups of neurons and neural pathways. Central to this mechanism is prolonged activation of the brain’s microglia, a major source of immune cytokines, chemokines, and other immune factors as well as the excitotoxins, glutamate, aspartate and quinolenic acid.
Inflammation in the brain, especially if prolonged, triggers excitotoxicity, which over time destroys, first synaptic connections and then eventually neuronal cell bodies — something seen in postmortem examinations of autistic brains. Associated with immunoexcitotoxicity one also witnesses, as an intimate part of the process, high levels of reactive oxygen and reactive nitrogen species (ROS/RNS) and lipid peroxidation products accumulating within these affected brain areas. This has also been confirmed in cases of schizophrenia.
Both postmortem and in vivo Positron emission tomography (PET) scanning have demonstrated microglial activation in the affected areas of the brain of schizophrenia patients and autism spectrum disorder (ASD) patients. Interestingly, the areas of the brain most affected in schizophrenia have been shown to have higher densities of microglia than are normally seen in healthy brains (28% higher in frontal cortex and 57% higher in temporal area). We see this same finding in Parkinson’s disease, with the substantia nigra having the highest density of microglia in the brain. In the normal brain, there are relatively high concentrations of microglia in the limbic areas of the brain and prefrontal cortex.
Microglia priming is also a critical process in this disorder and most neurodegenerative diseases. This is a state where microglia have a dramatic increase in the enzyme systems and cellular signaling responsible for producing the destructive elements released by fully active microglia, such as the excitotoxins and immune elements. Despite this alteration, these destructive elements are not released by the primed microglia. But, once fully activated, they are released and are significantly more destructive than one sees with the usual activated microglia. Systemic inflammation is a major factor in microglial priming, but a number of environmental factors, both internal and external, can prime microglia. Aging itself primes microglia. Fully activated microglia have activation of neuronal and inducible nitric oxide synthetase (nNOS and iNOS) as well as COX-2 production of proinflammatory prostaglandins (PGE2).
Activation of microglia in schizophrenia was first reported in 1999, where microglial activation was seen in the frontal cortex and hippocampus in 14 patients. Since then, this has been confirmed in a number of studies. The hypoglutaminergic theory of schizophrenia is based on the findings that many of the symptoms of schizophrenia improve significantly following stimulation by NMDA receptor agonist and that specific glutamate blocking drugs (PCP and ketamine) can induce a schizophrenia-like condition. The problem I find with this theory is that early in the course, even before the obvious symptoms develop, one sees subtle psychological changes, indicating that the process begins much earlier and is not fully manifest until a great deal of destructions occurs in brain connectivity and neuronal loss. In essence, early in the course of the disease one witness excitotoxicity, which later, because of destruction of both astrocytes and glutamatergic neurons, results in brain glutamate levels falling. A loss of these neural pathways leads to further brain dysfunction, especially in memory. It should be kept in mind that glutamate is the major transmitter for over 50% of the brain and 90% of the cortex.
It is also important to appreciate that schizophrenia and autism spectrum disorders share core symptoms and overlap in many ways pathologically, mainly by extensive microglial activation and similar behavioral attributes. In addition, there appears to be a strong genetic link associated with both and, interestingly, a number of these genes have to do with control of microglial function (TREM2, TLRs, TYRO proteins, etc). It has also been shown that success in treatment parallels lowering of these inflammatory cytokines. Since, at this stage, glutaminergic neurons are already depleted and dysfunctional in many brain areas, further lowering of glutamate may be counterproductive clinically. Yet, there is some evidence that excitotoxicity continues in some brain areas and that metabotrophic glutamate receptor 5 (mGluR5) is overactive (which is excitotoxic). A more selective suppression of mGluR5 may be beneficial.
It is also known that the GluR2 lacking AMPA receptors*, which are calcium permeable, is upregulated during inflammatory states and has been linked to schizophrenia. Another link between excitotoxicity and inflammation is the effect of inflammation on serotonin metabolism, principally the kynurenine pathway, with the generation of the excitotoxin product quinolenic acid (QUIN). QUIN activates the NMDA receptor. Brain and CSF levels of QUIN are increased in schizophrenia and autism spectrum disorders. It should also be appreciated that there is an intimate connection and interaction between glutamate receptors and other neurotransmitter receptors, such as dopamine, acetylcholine and serotonin.
It was noted in one of the comments that . It is known that nicotine is a powerful stimulant of the alpha-7 nicotinic acetylcholine receptors, which are suppressors of inflammation and generally responsible for controlling inflammation within the brain — the so-called cholinergic anti-inflammatory system. A loss of nicotinic receptors, which occurs in schizophrenia and Autism Spectrum Disorders (ASDs), is thought to enhance brain inflammation. The anti-inflammatory cytokine, TGF-ß1 is also severely lowered in schizophrenia and ASD. So, we see a serious imbalance between proinflammatory mechanisms and anti-inflammatory mechanisms within the schizophrenic brain, with proinflammatory predominance.
The big question is: What is causing the chronic, low level inflammation? It has been shown clearly that prenatal infections in the mother can cause postnatal schizophrenia and more recent studies using non-infectious Poly:IC have shown that the mechanism involves cytokine elevations, principally IL-6 and not actual infection. In animal models, blocking IL-6 (and IL-1ß) can prevent the schizophrenia/ASD onset postnatally.
Another link that has a lot of validity, certainly in some cases of both ASD and schizophrenia, is gut inflammation. Gliadin and gluten have been shown to trigger chronic microglial activation and is linked clinically to a number of cases of schizophrenia. Several reports describe significant improvement of cases upon assuming a gluten-free diet, while others found little or no improvement. There may be an explanation for the failure of improvement on a gluten free diet in these cases. First, it should be appreciated that these gluten-linked cases are associated with a non-celiac gluten sensitivity, which does not show the usual antibody profile of typical celiac disease cases and there is no villus atrophy on duodenal biopsy. This has been called Non-Celiac Gluten Sensitivity (NGCS).
As for why some cases fail to improve, it has been shown that gluten can trigger increased gut permeability, which can persist in some cases after starting a gluten-free diet, as there are often other contributing factors also linked to gut permeability, such as use of nonsteroidal anti-inflammatory drugs (NSAIDs). Translocation of other food proteins and colon/intestinal bacteria can trigger continued microglial activation with resulting persistent immunoexcitotoxicity. In addition, gut inflammation can send afferents via the vagus nerve that activate brain microglia. In addition, once the microglia are primed, other environmental factors can precipitate continued microglial activation, such as heavy metals, aluminum, fluoroaluminum, microparticulate fuels and certain pesticides/herbicides.
It is also of interest that many of the antipsychotic medications used to treat schizophrenia are known to suppress microglial activation and alter glutamate receptor function. These include risperidone, clozapine and olanzapine. Some also reduce reactive oxygen species damage (ROS). Studies have shown that drug responses correlate with lowering of S100B levels, a marker for brain inflammation.
As for the neurotransmitters, especially dopamine, it has been shown that treatment resistant forms of schizophrenia (type B patients) were associated with relatively normal levels of dopamine synthesis in the striatum and elevated glutamate levels in the anterior cingulate cortex. Lowering of the excitotoxin QUIN has also been shown to be related to clinical improvement.
The disruption of several neurotransmitters in schizophrenia is consistent with immunoexcitotoxicity, as a number of neuron types and receptor types and subtypes are affected by high levels of inflammation and excitotoxicity, with associated elevations in reactive oxygen and nitrogen species and lipid peroxidation — that is, these changes are epiphenomenon. In my opinion, we should be addressing the central mechanism of the problem (immunoexcitotoxicity and microglial activation) rather than attempting to fine tune neurotransmitter disruptions, which can appear in a complex, variable and often confusing presentation. This also requires attention to gut inflammation and correction of the microbiome. I would refer the reader to my recent paper in the journal Surgical Neurology International in which I describe in detail .
Footnote
* The AMPA is an α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, an ionotropic transmembrane receptor for glutamate that mediates fast synaptic transmission in the central nervous system (CNS) and it is separate from an NMDA receptor.
Written by Russell L. Blaylock, MD
Dr. Russell L. Blaylock is President of Advanced Nutritional Concepts and Theoretical Neurosciences in Jackson, Mississippi. He has written numerous path-blazing scientific papers and many books, including Excitotoxins — The Taste That Kills (1994), Bioterrorism: How You Can Survive (2001), Health and Nutrition Secrets (2002), Prescriptions for Natural Health (2016), Cellular and Molecular Biology of Autism Spectrum Disorders, co-authored with Anna Strunecka, Mark Hyman and Ivo Paclt (2010), and Natural Strategies for Cancer Patients (2019). He is Editor-in-Chief of the Neuroinflammatory Section of Surgical Neurology International (SNI).
This article may be cited as: Blaylock RL. Inflammation, immunoexcitotoxicity and microglia activation in schizophrenia. HaciendaPublishing.com, January 31, 2018 Available from: https://haciendapublishing.com/inflammation-immunoexcitotoxicity-and-microglia-activation-in-schizophrenia-by-russell-l-blaylock-m-d/
Copyright ©2018 Hacienda Publishing Inc.